

Research Interests
-
Research Interests
In the Whiteley Lab, we are interested in the social lives of bacteria. Currently we are utilizing new technologies combined with classical genetic techniques to address questions about microbial physiology, ecology, virulence, and evolution. In particular we are working on tackling the following questions:
- How do bacteria communicate?
- How do polymicrobial interactions impact physiology and virulence?
- How do bacteria evolve in the host?
- How do nutrients available at infection sites impact behavior and virulence?
With a large variety of resources at UT Austin we are taking advantage of several unique opportunities for collaboration. With the Shear lab in the chemistry department we are using multiphoton lithography and confocal microscopy to study small microbial communities in “bacterial lobster traps” that often mimic the sizes of populations observed in nature more accurately than traditional, dense batch culture techniques. With the UT Genome Sequencing and Analysis Facility and the Texas Advanced Computing Center we are harnessing high-throughput sequencing methods including RNA sequencing (RNA-Seq) and transposon sequencing (Tn-Seq)
-
RNA-Seq
Method
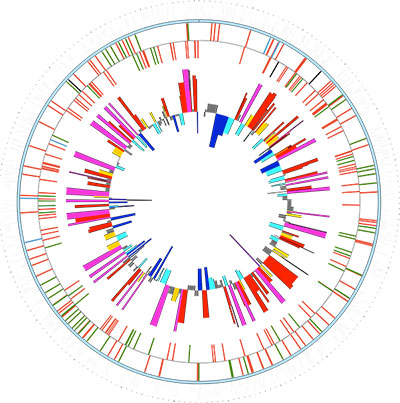 High-throughput RNA sequencing (RNA-Seq) is a relatively new technology used to study the transcriptomics of organisms and communities. Similar to microarray analysis, RNA-Seq can be used to identify differentially expressed genes under different conditions; however, RNA-Seq can also be used to identify transcriptome architecture, including transcriptional start sites for genes, polycistronic operons, and novel non-coding RNAs. To carry out RNA-Seq, RNA is isolated from a sample, it is reverse transcribed to generate a cDNA library, and the cDNA is sequenced using the Illumina HiSeq.
High-throughput RNA sequencing (RNA-Seq) is a relatively new technology used to study the transcriptomics of organisms and communities. Similar to microarray analysis, RNA-Seq can be used to identify differentially expressed genes under different conditions; however, RNA-Seq can also be used to identify transcriptome architecture, including transcriptional start sites for genes, polycistronic operons, and novel non-coding RNAs. To carry out RNA-Seq, RNA is isolated from a sample, it is reverse transcribed to generate a cDNA library, and the cDNA is sequenced using the Illumina HiSeq.
Approach
For years we have known that polymicrobial infections are often difficult to treat due to synergy. Polymicrobial synergy refers to interactions between microbes that achieve a result greater than the sum of the microbes in isolation. We have identified synergistic interactions between the Gram-negative periodontal pathogen Aggregatibacter actinomycetemcomitans (Aa) and oral streptococci. While we know that this synergy is at least partially mediated by lactate and hydrogen peroxide, it is unknown how global genetic mechanisms impact this interaction. We are using RNA-Seq to identify the genes underlying synergy in a model polymicrobial community including Aa and S. gordonii. Additionally, we are building upon these initial studies using RNA-Seq to study community structure and function in real human oral infections, polymicrobial wound infections, and Cystic Fibrosis lung infections.
-
Genome Sequencing
Method
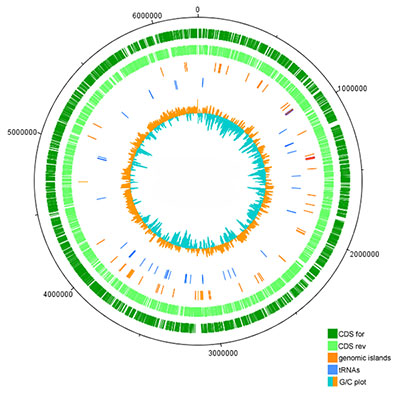
High-throughput DNA sequencing can determine the genetic content of a microbe in less than a week’s time. This powerful technology allows us to study individual microbes and microbial communities. This simple process begins by isolating total DNA from a sample, including chromosomal, plasmid, and phage DNA. The isolated DNA is sheared into small fragments that are amenable to library preparation. After shearing, sequencing adapters are ligated onto DNA fragments and sequenced using the Illumina MiSeq.
Approach
It is appreciated that chronic infections are exacerbated by microbial adaptation to the host environment; however, the precise mutations underlying adaptation remain elusive. We are utilizing next generation genome sequencing to study in vivo evolution of Pseudomonas aeruginosa during chronic Cystic Fibrosis (CF) lung infections. Previously, using microarray analysis, we identified 24 P. aeruginosa genes under natural selection during chronic CF infections. To determine the genetic basis for this adaptation, we are using whole genome sequencing to generate an evolutionary history of multiple infectious P. aeruginosa lineages during 1000s of generations inside the host. Ultimately we will use this history to identify the genetic mutations that benefit the microbe during infection.
-
Tn-Seq
Method
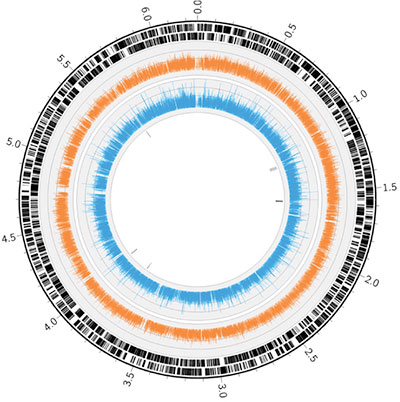
Transposon sequencing (Tn-Seq) is a tool to globally profile the location and frequency of transposons in an insertion mutant pool. Tn-Seq experiments are similar to classic transposon mutant screens, but instead of screening mutants individually, a mutant pool is investigated en masse through use of next-generation sequencing. When the mutant pool is grown under a selective condition, its profile is altered in that susceptible mutants are depleted from the pool. Tn-Seq can then be used to compare the input and output populations to rapidly identify the comprehensive set of genes required to survive under that condition. With mutant pools at high enough saturation, Tn-Seq can also identify important intergenic regions (e.g. promoters) and even sub-genetic regions (e.g. conserved motifs) that are required for fitness. Procedures for sequencing a mutant pool involve capturing the genomic DNA flanking insertion sites prior to deep sequencing on the Illumina HiSeq or MiSeq.
Approach
To successfully colonize and proliferate within a host organism, pathogens must overcome a variety of restrictions imposed by the host environment. Pathogens have evolved mechanisms to respond rapidly to environmental changes (e.g. acid or osmotic shock), acquire multiple carbon sources, and resist antimicrobial factors (e.g. produced endogenously by the host or supplied through antibiotic treatment). These mechanisms can each contribute to infection. We are interested in applying Tn-Seq to learn about mechanisms that enable the model pathogens Pseudomonas aeruginosa (Pa) and Aggregatibacter actinomycetemcomitans (Aa) to survive these stresses. Pa is an infamous pathogen of the cystic fibrosis lung and chronic wounds, and Aa is an implicated causative agent of periodontitis. These infections are often polymicrobial, and we have previously identified interspecies interactions that may worsen infection during co-colonization with Pa and Staphylococcus aureus and Aa and Streptococcus gordonii. We are using Tn-Seq to establish genetic interaction networks during growth of Pa and Aa under a suite of in vitro stresses and within polymicrobial communities. These experiments will inform results of Tn-Seq experiments performed using both mono-culture and polymicrobial animal infection models.
-
SECM
Method
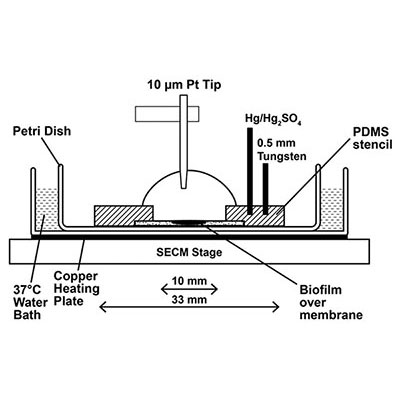 Scanning electrochemical microscopy (SECM) is an analytical tool capable of making spatially resolved concentration measurements of redox-active small molecules over the surface of a biological substrate. SECM offers the power to set the distance between a substrate, such as a bacterial cell or a biofilm, and the ultramicroelectrode sensing tip (≤10 μm) with nanometer resolution in three dimensions using a feedback approach curve1. Once positioned, the tip can directly measure the local concentration and redox state of small molecules surrounding a bacterial population in real time. The potential of the tip is controlled via computer, and can be changed to probe the concentration and redox status of multiple different electroactive molecules throughout an experiment. Scanning the tip in the x-y direction creates a spatially resolved reactive map of a surface. Since this method provides quantitative information in real time, monitoring a biological substrate over time allows SECM to generate a spatio-temporal profile of a dynamic environment.
Scanning electrochemical microscopy (SECM) is an analytical tool capable of making spatially resolved concentration measurements of redox-active small molecules over the surface of a biological substrate. SECM offers the power to set the distance between a substrate, such as a bacterial cell or a biofilm, and the ultramicroelectrode sensing tip (≤10 μm) with nanometer resolution in three dimensions using a feedback approach curve1. Once positioned, the tip can directly measure the local concentration and redox state of small molecules surrounding a bacterial population in real time. The potential of the tip is controlled via computer, and can be changed to probe the concentration and redox status of multiple different electroactive molecules throughout an experiment. Scanning the tip in the x-y direction creates a spatially resolved reactive map of a surface. Since this method provides quantitative information in real time, monitoring a biological substrate over time allows SECM to generate a spatio-temporal profile of a dynamic environment.Approach
Bacteria use chemical signals to communicate and modulate gene expression as a group in a process called quorum sensing (QS). Additionally, cells recognize and respond to many other small molecules to adapt and survive in nature. We are working in collaboration with Prof. Allen Bard’s group at UT Austin to characterize these behaviors in complex, dynamic bacterial environments using SECM (a schematic of the SECM apparatus is shown below2) We have employed SECM to spatially map hydrogen peroxide production in mixed species biofilms containing Streptococcus gordonii and Aggregatibacter actinomycetemocomitans3, and demonstrated that Pseudomonas aeruginosa biofilms produce and maintain an “electrocline” (a gradient of redox potential) of pyocyanin, a QS signal and antimicrobial molecule, in its reduced form surrounding the biofilm2. We continue to work together with the Bard group to use SECM to probe bacterial interactions among small populations of bacteria and individual cells.
-
Bacterial Lobster Traps
Method
 Mask-based multiphoton lithography (MPL) is a versatile microfabrication approach developed in the Shear Group that has the ability to pattern porous hydrogel materials with sub-micron spatial resolution in three dimensions. In this direct-write fabrication method, solid structures are formed from aqueous protein precursors via covalent crosslinking reactions between amino acid residues in the focal volume of a pulsed laser beam. Complex three-dimensional microstructures can be formed layer-by-layer by translating the focus of the scanning beam in the optical (z) axis through solution. In collaboration with Prof. Jason Shear’s laboratory at UT Austin, we employ this highly adaptable fabrication platform to create customized microstructures to house small bacterial populations and use microscopy (light and confocal) to monitor group behaviors in real time.
Mask-based multiphoton lithography (MPL) is a versatile microfabrication approach developed in the Shear Group that has the ability to pattern porous hydrogel materials with sub-micron spatial resolution in three dimensions. In this direct-write fabrication method, solid structures are formed from aqueous protein precursors via covalent crosslinking reactions between amino acid residues in the focal volume of a pulsed laser beam. Complex three-dimensional microstructures can be formed layer-by-layer by translating the focus of the scanning beam in the optical (z) axis through solution. In collaboration with Prof. Jason Shear’s laboratory at UT Austin, we employ this highly adaptable fabrication platform to create customized microstructures to house small bacterial populations and use microscopy (light and confocal) to monitor group behaviors in real time.Approach
Structures made using protein MPL have facilitated phenotypic studies of small aggregates of cells. A single bacterium can be captured within a picoliter-sized container. These porous bacterial “lobster traps,” facilitate normal cell growth, which can be observed using standard light and confocal microscopy techniques One cell enclosed in a three-dimensional user-defined geometry can become a clonal population quickly. We have studied growth kinetics, bacterial cell-cell communication (quorum sensing), and antibiotic resistance phenotypes of small, densely packed populations. Currently, we aim to characterize how sensing small molecules, such as nutrients (e.g., O2) and metabolic byproducts (e.g., H2O2) impact microbial behavior in high density, low cell number environments.
